
Unbiased artificial chemical shift data base
The unbiased artificial chemical shift data base (UACSB) was recalculated from the unbiased protein structural data base NH3D (Sillitoe et al., 2013). The structures with added protons were energy minimized in explicit water using the molecular dynamics program GROMACS (version 4.6.1) (Berendsen et al. 1995; Hess et al. 2008). The final energy minimized structure of each model was then used for a molecular dynamics simulation in explicit water. After equilibration, 400 ps trajectories have been calculated. 11 equidistant structures were extracted from these trajectories for creation of the UACSB database. Since stereospecifically defined atom names may change after energy minimization (bond rotation), it was checked again and if necessary corrected after minimization with the AUREMOL (Gronwald and Kalbitzer, 2004) tool IUPACify. Chemical shifts were calculated using SHIFTS (version 4.3) (Osapay and Case, 1991) or SHIFTX2 (version 1.04) (Neal et al., 2003). For details see Harsch et al. (2017).
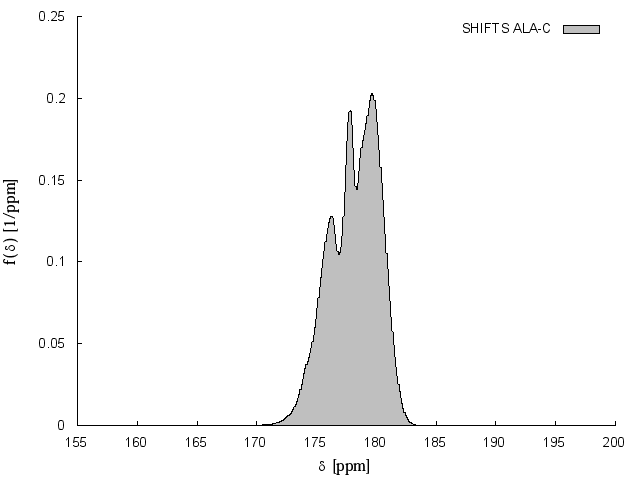
Display UACSB chemical shift distribution for a given atom in a given amino acid:
When using the database please cite:
Harsch, T., Schneider, P., Kieninger, B., Donaubauer, H., Kalbitzer, H. R. (2017) Stereospecific assignment of the asparagine and glutamine side chain amide protons in proteins from chemical shift analysis. J. Biomol. NMR 67, 157-164.
References
- Berenden, H.J.C., van der Spoerl, D, van Dunen, R. (1995) GROMACS: A message-passing parallel dynamics impmementation. Comp. Phys. Comm. 91, 43-56.
- Gronwald, W., Kalbitzer, H.R. (2004) Automated Structure Determination of Proteins by NMR Spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 44, 33-96.
- Harsch, T., Schneider, P., Kieninger, B., Donaubauer, H., Kalbitzer, H. R. (2017) Stereospecific assignment of the asparagine and glutamine side chain amide protons in proteins from chemical shift analysis. J. Biomol. NMR 67, 157-164.
- Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E. (2008): GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4 (3), 435-447
- Neal, S., Nip, A.M., Zhang, H., Wishart, D.S. (2003) Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR 26, 215 -240
- Osapay, K., Case, D.A. (1991) A new analysis of proton chemical shifts in proteins. J. Am. Chem. Soc. 113, 9436-9444.
- Sillitoe, I., Cuff, Alison L. (2013) New functional families (FunFams) in CATH to improve the mapping of Conserved functional sites to 3D structures. Nucleic Acids Res, 2013. 41 D490-D498.