
AUREMOL is a software package for automated NMR spectrum evaluation and protein structure determination, compatible with AURELIA. It has been developped and is still being improved at the University of Regensburg in cooperation with Bruker Biospin GmbH and Kalbitzer Innovations UG.
When using the program please cite this site and/or
Gronwald, W. and Kalbitzer, H. R. (2004) Automated Structure Determination of Proteins by NMR Spectroscopy. Progr. NMR Spectr. 44, 33-96.
Features
Display of multidimensional NMR spectra
Postprocessing of NMR spectra
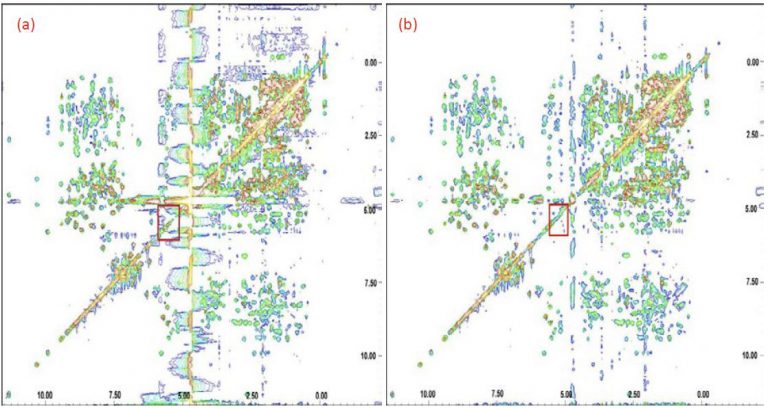
- Water signal suppression by SSA
- Spectral improvement by ICA
- Automated base line correction
Automated adaptive peak picking in n-dimensions
- Threshold-based peak picking
- Adaptive peak picking
- Bayesian signal recognition
Automated assignment of the NOESY cross peaks
- Optimization of chemical shift lists for a given nD-spectrum
- Probabilistic assignments of NOESY cross peaks from a distance data base
Simulation of 2D NOESY and 3D NOESY-HSQC spectra using the complete relaxation matrix formalism
- Homo- and heteronuclear dipolar relaxation
- Relaxation via chemical shift anisotropy
- Various spectral density functions including anisotropic tumbling and internal motions
- Calculation of individual T2-values
- Simulation of J-couplings
Calculation of interatomic distances and error intervals using the complete relaxation matrix formalism
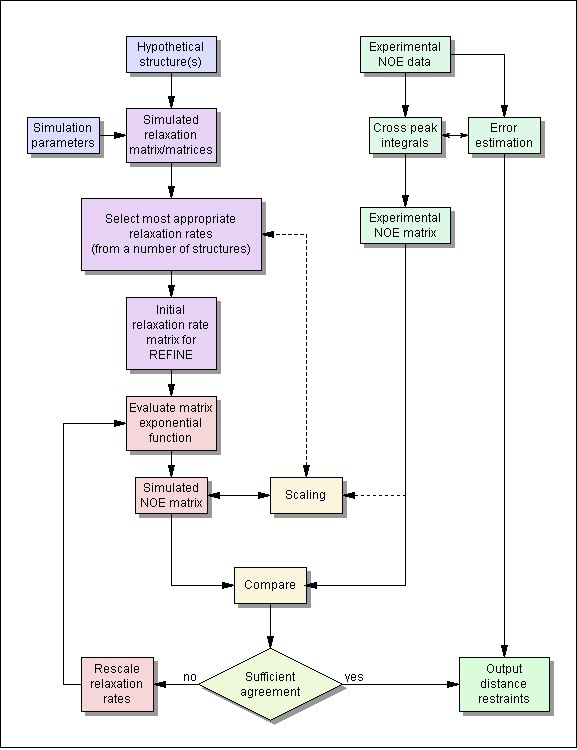
- Automated volume integration by iterative segmentation including volume error calculation
- Automated calculation of interatomic distances from the relaxation matrix
- Consideration of internal motions
- Calculation of indivudal distance errors
NMR structural refinement
- Refinement of NMR structures by x-ray structures of homologeous proteins
- Calculation of substitute restraints
Calculation of NMR-R-factors
- Problem specific definitions of R-factors
- Calculation of global and local R-factors
- Calculation of the R-factors on the basis of the complete relaxation matrix formalism
Interaction of (small) ligands with proteins and identification of binding sites
- Simulated annealing based homology modeling of proteins and protein complexes
- Calculation of combined chemical shifts and definition of binding sites
- Automated assignment of HSQC spectra from NOESY data and x-ray structures
NMR based quality control of biomacromolecules (AUREMOL-QTA)
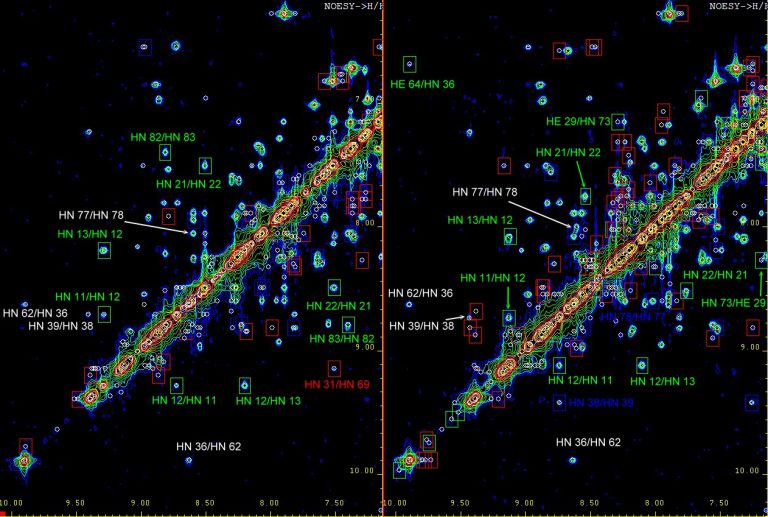
- Automated comparison between nD-spectra of the test sample with (a set of) reference spectra of a protein
- Identification of partial denaturation
- Identification of protein modifications
- Quality control of small molecules
Automated analysis of spectral changes induced by external perturbations
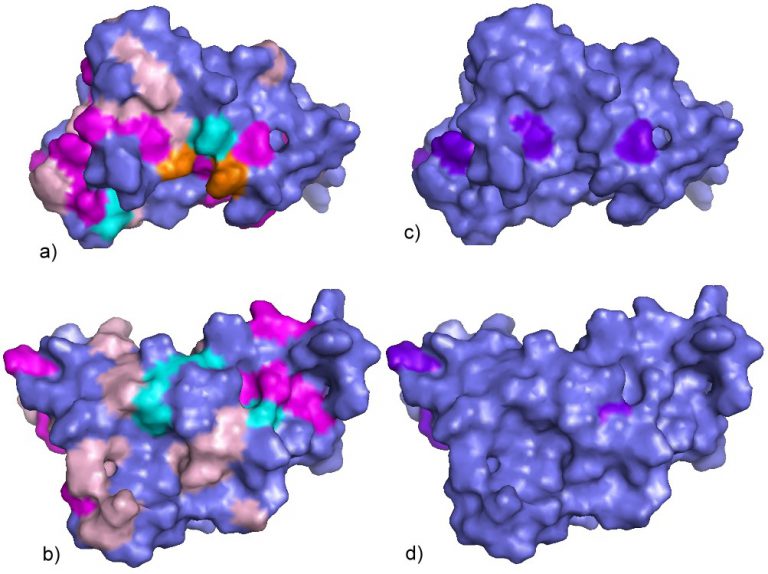
- Automated identification of residues perturbed by pressure changes, temperature changes or ligand binding
- Identification on the basis of chemical shift changes
- Identification on the basis of cross peak volume changes
- Identification on the basis of T2-changes
- Identification on the basis of combined effects
Acknowledements
The program AUREMOL is a successor of the program AURELIA (Neidig et al. 1995) developped in the group of H. R. Kalbitzer at the Max-Planck-Institute for Medical Research, Heidelberg, Germany. Many routines for data processing and data evaluation were developped at this time (see References below) and are also part of AUREMOL. AUREMOL was mainly written in the group of Prof. H. R. Kalbitzer at the University of Regensburg in cooperation with Dr. K. P. Neidig (Bruker Biospin).
The following members of the group contributed to AUREMOL:
Prof. Dr. Wolfram Gronwald
Dr. Konrad Brunner
Dr. Andre Fischer
Dr. Jochen Trenner
Dr. Renate Kirchhöfer
Dr. Adel Nasser
Dr. Bernhard Ganslmeier
Dr. Adrian Görler
Andreas Ried
Josef H. Scheiber
Dr. Kumaran Baskaran
Dr. Silvia DeSanctis
Dr. Massimo Wilhelm Maloni
Dr. Bärbel Kieninger
Dr. Harald Donaubauer
Dr. Tobias Harsch
References and related publications
Computer-aided assignment
- Neidig, P., Bodenmüller, H. & Kalbitzer, H. R. (1984)
Computer Aided Evaluation of Two-Dimensional NMR Spectra of Proteins. Biochem. Biophys. Res. Comm. 125, 1143-1150. - Glaser, S. & Kalbitzer, H. R. (1987)
Automated Recognition and Assessment of Cross Peaks in Two-Dimensional NMR Spectra of Macromolecules. J. Magn. Reson. 74, 450-463. - Groß, K.-H. & Kalbitzer, H. R. (1988)
Distribution of Chemical Shifts in 1H Nuclear Magnetic Resonance Spectra of Proteins. J. Magn. Reson. 76, 87-99. - Neidig, K.-P., Saffrich, R., Lorenz, M. & Kalbitzer, H. R. (1990)
Cluster Analysis and Multiplet Pattern Recognition in Two-Dimensional NMR spectra. J. Magn. Reson. 89, 543-552. - Kalbitzer, H. R., Neidig, K.-P., Geyer, M., Saffrich, R. & Lorenz, M. (1991)
Pattern Recognition in Two-Dimensional NMR Spectra of Proteins. In ‘Computational Aspects of the Study of Biological Macromolecules by Nuclear Magnetic Resonance´ (J.C. Hoch, ed.) pp 175-190, Plenum Press, New York. - Kalbitzer, H.R. (1994)
Computer- Aided Analysis of Multidimensional NMR Spectra. In “Two-dimensional NMR-Spectroscopy: Applications for Chemists and Biochemists” (W.R.Croasmun und R.Carlson, eds) pp 581-618, Verlag Chemie, Weinheim. - Neidig, K.-P., Geyer, M., Görler, A., Antz, C., Saffrich, R., Beneicke, W., & Kalbitzer, H. R. (1995)
AURELIA, a Program for Computer-Aided Analysis of Multidimensional NMR Spectra. J. Biomol. NMR. 6, 255-270. - Antz, C., Neidig, K.-P. & Kalbitzer, H. R. (1995)
A General Bayesian Method for an Automated Signal Class Recognition in 2D NMR Spectra Combined with a Multivariate Discriminant Analysis. J. Biomol. NMR. 5, 287-296. - Schulte, A. C., Görler, A., Antz, C., Neidig, K.-P. & Kalbitzer, H. R. (1997)
Use of Global Symmetries in Automated Signal Class Recognition by a Bayesian Method. J. Magn. Reson. 129, 165-172. - Gronwald, W., Moussa, S., Elsner, R., Jung, A., Ganslmeier, B., Trenner, J., Kremer, W., Neidig, K.-P., & Kalbitzer, H. R. (2002)
Automated assignment of NOESY NMR spectra using a knowledge based method (KNOWNOE). J. Biomol. NMR 23, 271-287. - Baskaran, K., Kirchhöfer, R., Huber, F., Trenner, J., Brunner, K., Gronwald, W., Neidig, K.-P. and Kalbitzer, H. R. (2009)
Chemical Shift Optimization in Multidimensional NMR Spectra by AUREMOL-SHIFTOPT. J. Biomol. NMR 43, 197-210.
Processing
- Glaser, S. & Kalbitzer, H. R. (1986)
Improvement of 2D NMR Spectra by Mean t1 Ridge Subtraction and Antidiagonal Reduction. J. Magn. Reson. 68, 350-354. - Neidig, K.-P. & Kalbitzer, H. R. (1988)
Improvement of 2D NMR Spectra by Matching Symmetry-Related Spectral Features. Magn. Reson. Chem. 26, 848-851. - Neidig, K.-P. & Kalbitzer, H. R. (1990)
Improved Representation of 2D NMR Spectra by Local Rescaling. J. Magn. Reson. 88, 155-160. - Mitschang, L., Neidig, K.-P. & Kalbitzer, H. R. (1990)
Suppression of Oscillatory Artifacts in Two-Dimensional NMR spectra. J. Magn. Reson. 90, 359-362. - Neidig, K.-P. & Kalbitzer, H. R. (1991)
Enhancement of Global Symmetries in Two-Dimensional NMR Spectra. J. Magn. Reson. 91, 155-164. - Saffrich, R. , Beneicke, W., Neidig, K.-P. & Kalbitzer, H. R. (1993)
Baseline Correction in n-Dimensional NMR Spectra by Sectionally Linear Interpolation. J. Magn. Reson. B 101, 304-308. - Beneicke, W. & Kalbitzer, H. R. (1994)
Data Filling in Two-Dimensional NMR Spectroscopy. J. Magn. Reson. A107, 134-140. - Geyer, M., Neidig, K.-P. & Kalbitzer, H. R. (1995)
Automated Peak Integration in Multidimensional NMR Spectra by an Optimized Iterative Segmentation Procedure. J.Magn.Reson.B 109, 31-38. - Maurer, T., & Kalbitzer, H. R. (1996)
Indirect Referencing of 31P and 19F NMR Spectra. J. Magn. Reson. B 113, 177-178. - Stadlhanner, K., Theis, F. J., Lang, E. W., Gronwald, W., Kalbitzer, H. R. (2003)
A matrix pencil approach to the blind source separation of artifacts in 2D NMR spectra. Neur. Inform. Process. 1, 103-110. - Stadlthanner, K., Tomé, A. M., Theis, F. J., Lang, E. W., Gronwald, W. & Kalbitzer, H. R. (2006)
Separation of water artefacts in 2D NOESY protein spectra using congruent matrix pencils. Neurocomp. 96, 497-522. - Böhm, M., Stadlthanner, K., Gruber, P., Theis, F. J., Lang, E. W., Tomé, A. M., Teixeira, A. R., Gronwald, W. & Kalbitzer, H. R. (2006)
On the use of simulated annealing to automatically assign decorrelated components in second-order blind source separation. IEEE Transact. Biomed. Engineer. 53, 810-820. - Malloni, W. M., De Sanctis, S., Tomé, A. M., Lang, E. W., Munte, C. E., Neidig, K. P. Kalbitzer, H. R. (2010)
Automated solvent artifact removal and base plane correction of multidimensional NMR protein spectra by AUREMOL-SSA. J. Biomol. NMR 47, 101-111. - De Sanctis, S., Malloni, W. M., Kremer, W., Tomé, A. M., Lang, E. W., Neidig, K. P. Kalbitzer, H. R. (2011)
Singular Spectrum Analysis for an Automated Solvent Artifact Removal and Baseline Correction of 1D NMR Spectra. J. Magn. Reson.210, 177-183. - De Sanctis, S., Malloni, W. M., Kremer, W., Lang, E. W., Kalbitzer, H. R. (2012)
Independent component analysis (ICA) and singular spectrum analysis (SSA) for solvent artifact suppression in one-dimensional NMR spectroscopy: a comparative analysis. Trends Appl. Spectr. 9, 1-15.
Protein-ligand and protein-protein interaction
- Schumann, F. H., Riepl, H., Maurer, T., Gronwald, W., Neidig, K.-P., & Kalbitzer, H. R. (2007)
Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275-289.
Quality control
- Gronwald, W., Kirchhöfer, R. Görler, A., Kremer, W., Ganslmeier, B., Neidig, K.-P. & Kalbitzer, H. R. (2000)
RFAC, a programm for automated NMR-R-factor estimation. J. Biomol. NMR 17, 137-151. - Gronwald, W., Brunner, K., Kirchhöfer, R., Trenner, J., Neidig, K.-P. & Kalbitzer, H. R. (2007)
AUREMOL-RFAC-3D, combination of R-factors and their use for automated quality assessment of protein structures. J. Biomol. NMR 37, 15-30. - Baskaran, K., Brunner, K., Munte, C. E., Kalbitzer, H. R. (2010)
Mapping of protein structural ensembles by chemical shifts. J. Biomol. NMR 48, 71-83.
Simulations
- Görler, A. & Kalbitzer, H. R. (1997)
RELAX: A Flexible Program for the Analysis of NOESY-Spectra by Backcalculation Based on the Complete Relaxation Matrix Formalism. J. Magn. Reson. 124, 177-188. - Görler, A., Gronwald, W., Neidig, K.-P. & Kalbitzer, H. R. (1999)
Computer Assisted Assignment of 13C or 15N edited 3D-NOESY-HSQC Spectra Using Back Calculated and Experimental Spectra. J. Magn. Reson. 137, 39-45. - Ried, A., Gronwald, W., Trenner, J. M., Brunner, K., Neidig, K.-P. & Kalbitzer, H. R. (2004)
Improved simulation of NOESY spectra by RELAX-JT2 including effects of J-coupling, transverse relaxation, and chemical shift anisotropy. J. Biomol. NMR 30, 121-131. - Möglich, A., Weinfurtner, D., Maurer, T., Gronwald, W. & Kalbitzer, H. R. (2005)
A restraint molecular dynamics and simulated annealing approach for protein homology modeling utilizing mean angles. BMC Bioinform. 6:91. - Möglich, A., Weinfurtner, D., Gronwald, W., Maurer, T. & Kalbitzer, H. R. (2005)
PERMOL: Restraint-based protein homology modeling using DYANA or CNS. Bioinform.21, 2110-2111. - Harsch, T., Dasch, C., Donaubauer, H., Baskaran, K., Kremer, W., Kalbitzer, H. R. (2013)
Stereospecific Assignment of the Asparagine and Glutamine Side Chain Amide Protons in Random-Coil Peptides by Combination of Molecular Dynamic Simulations with Relaxation Matrix Calculations. Appl. Magn. Reson.44, 319-331. - Harsch, T., Schneider, P., Kieninger, B., Donaubauer, H., Kalbitzer, H. R. (2017) Stereospecific assignment of the asparagine and glutamine side chain amide protons in proteins from chemical shift analysis. J. Biomol. NMR 67, 157-164.
Structure determination
- Gronwald, W. and Kalbitzer, H. R. (2004)
Automated Structure Determination of Proteins by NMR Spectroscopy. Progr. NMR Spectr. 44, 33-96. - Gronwald, W., Brunner, K., Kirchhöfer, R., Nasser, A., Trenner, J., Ganslmeier, B., Riepl, H., Ried, A., Schreiber, J., Elsner, R., Kalbitzer, H. R. & Neidig, K.-P. (2004)
The new AUREMOL program. Automation of protein structure elucidation. Bruker Report 154/155, 11-14. - Brunner, K., Gronwald, W., Trenner, J. M., Neidig, K.-P. & Kalbitzer, H. R. (2006)
A General Method for the Unbiased Improvement of Solution NMR Structures by the Use of Related X-Ray Data, the AUREMOL-ISIC Algorithm. BMC Struct. Biol. 346, 301-305. - Cano, C., Brunner, K., Baskaran, K., Elsner, R., Munte, C. E. and Kalbitzer, H. R. (2009)
Protein structure calculation with data imputation: the use of substitute restraints. J. Biomol. NMR 45, 397-411. - Gronwald, W. and Kalbitzer, H. R. (2010)
Automated protein NMR structure determination in solution.In “Computational Biology” (D. Fenyo, ed) Humana Press, Chapter 7, 95-127.