
The program PERMOL+ is part of the software package AUREMOL but can be used also as standalone version. It was developed to extract information about the structure(s) of known protein(s) in the form of restraints – specifically distances between atoms, dihedral angles and H-bonds – and then transfer this information to an unknown protein. These restraints can be used for molecular dynamics as restrictions of the configuration space. The ansatz also allows to include additional experimental restraints as they are usually obtained by NMR-spectroscopy (interatomic distances from NOEs or chemical shift perturabations, dihedral angles from J-couplings or chemical shifts, orientational information from residual dipolar couplings, H-bonds from an analysis of interatomic distances, exchange rates or long-range couplings from HSQC-spectroscopy. Whereas the earlier version PERMOL (Möglich et al., 2005a,b) was only able to predict structures of a single protein molecule, PERMOL+ (Kieninger, 2015) is able to preduct structures from protein complexes with high reliability.
+
Homology Modeling of Proteins and Protein complexes
Workflow of Homology Modeling with PERMOL+
PERMOL was originally developed for homology modeling, which is based on the observation that two proteins share the same folding pattern even if they match in only 20 % of their sequence. This technique implicates three main steps: First alignment of the unknown protein to one or more models, then the generation of the restraints – here the user has a lot of possibilities e.g. the selection of the atom types or the upper und the lower limit of the distance between the atoms, which should be considered at generation, or the selection of only parts of the sequence, and finally calculation of the new structure with the molecular dynamic programs XPLOR or CNS.
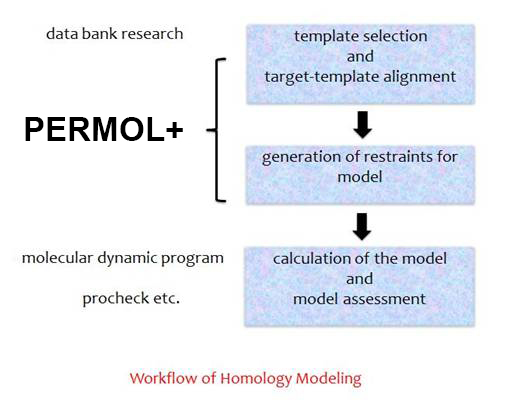
How to create restraints for the prediction of structures of proteins or protein complexes with PERMOL+?
Step 1:
Use AUREMOL or download the standalone version of PERMOL+. Start the program.
Step 2:
Feed target sequence(s) of the protein(s). You will get the model: One-letter-code or an AUREMOL sequence file or the possiblity to extract it from a PDB file.
Step 3:
Prepare the PDB files with your homologous proteins from which the informations will be extracted: Add hydrogens (if you will use XRAY-structures) and use IUPAC names for the atoms! Check the stereochemical nomination, too, because if there are mistakes you get structures with a lot of violations und big terms of energy!
Step 4:
By the generation of restraints you choose between the standard set of restraints (see figure) or a user defined one!
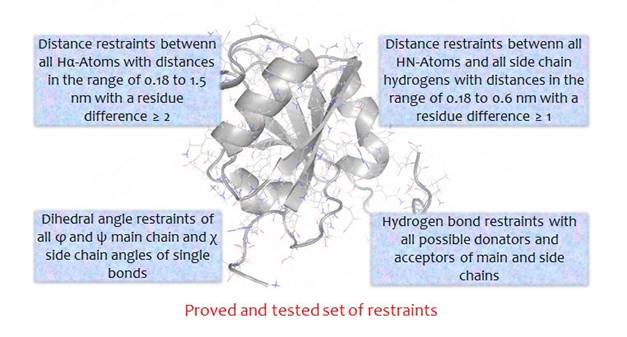
Step 5:
Choose a path for output!
Step 6:
After you have clicked the OK-Button the alignment will be shown. This alignment can be edited, but the sequences cannot be modified at this step. However, you can shift the gaps (-). Be careful that at the end of your editing the sequences must have the same length: The signs + at the beginning and the end of the sequences must align. In addition you have the option to exclude parts of the sequences from information generation.
Step 7:
Then the restraints will be generated.
Structure calculation
Now you can use these restraints to calculate structures with CNS (Brünger et al., 1998; http://structure.usc.edu/cns/main/frame.html). Thereby you have to pay attention the following points:
• Because of the big number of generated restraints (much more than by experimental data analysis) you have to adapt the possible parameters in file readdata (NMR module) to read in all restraints. By the final water refinement according to Linge et al. (2003) you have to change the parameter nrestraints.
• To optimize the weighting between the potentials which represent the a priori knowledge of proteins and these potentials which use the informations from the with PERMOL+ generated restraints by structure calculation you must decrease the parameters md.hot.noe, md.hot.cdih, md.cool.noe, md.cool.cdih, md.cart.noe, md. cart.cdih, md.pow.noe and md.pow.cdih in script anneal.inp. In the water refinement script these parameters have to change together with the parameter scale. The right choice of parameters can be detected by small values of the energies obtained – especially of the energies which represent the physical and chemical properties of proteins such as interatomic bond length or Van-der-Waals energies of the generated structures, what means that the covalent structures will be influences, but not be deformed by the PERMOL+ restraints.
• The time steps md.hot.ss, md.cool.ss, md.cart.ss and md.pow.ss must be smaller and – adapted to this – the number of iteration steps md.hot.step, md.cool.step, md.cart.step and md.pow.step have to increase. Otherwise it is possible that atoms „get lost“ during the simulation, if the forces affecting these atoms are too strong because of the big number of restraints, and then in one of the steps of iteration their new place could be out of the possible volume.
Acknowledgements
The new version of PERMOL+ unifies ideas from: Andreas Möglich, Daniel Weinfurtner, Till Maurer, Wolfram Gronwald, Josef Scheiber, Konrad Brunner, Carolina Cano, Michael Ebel, Bärbel Kieninger and Hans Robert Kalbitzer.
Authors of this new version are Bärbel Kieninger and Hans Robert Kalbitzer.
References
Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros , P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu , N. S., Read, R. J., Rice , L. M, Simonson , T., Warren, G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. A. Crystal. D Biol. Crystallogr. 54, 905-921.
Cano, C., Brunner, K., Baskaran, K., Elsner, R., Munte , C. E., Kalbitzer , H. R. (2009) Protein structure calculation with data imputation: the use of substitute restraints. J. Biomol. NMR 45, 397-411.
Kieninger, B. (2015) Entwicklung von Methoden zur Strukturvorhersage von Proteinkomplexen und zur Untersuchung von Proteinzuständen mit Hochdruckkernspinresonanzspektroskopie. Dissertation, University of Regensburg.
Möglich, A., Weinfurtner, D., Gronwald, W., Maurer, T., Kalbitzer, H. R. (2005a) PERMOL: restraint-based protein homology modeling using DYANA or CNS. Bioinformatics, 21 (9). 2110-2111.
Möglich, A., Weinfurtner, D., Maurer, T., Gronwald, W., Kalbitzer, H. R. (2005b) A restraint molecular dynamics and simulated annealing approach for protein homology modeling utilizing mean angles. BMC Bioinformatics, 6:91.